DISCLAIMER: All images on this document are not mine, and have their rights reserved by their owners.
In this post i'll present the study I made for a state of art project for my class in this semester, where the subject of research was the field of protein structure prediction.
Here is the Zotero link with the papers that I found, and other links: https://www.zotero.org/rdenadai/collections/6WDHP79S
Protein Structure Prediction is another biggest challenge that mankind is looking to solve. If solved or at least partially solved it could revolutionize medicine and drug industry.
At the end of the day, the search is the simple process of given a amino acid sequence, one must predict the 3D structure of the protein given physical restrictions (lower energy cost given by Gibbs Free Energy).
Motivation
Proteins are a important part of every organism, they, for example, help in the transport of molecules between cells. Given their importance, as much proteins we know, the best we could find, for example, better treatments to diseases.
In this sense I leave here the link about the excellent project done by my classmates, where they investigate the relationship between human proteins that interact with SARS-COV-2.
Research
I start the research choosing terms to search for and what bases i'll apply those terms.
Not an easy task to find terms to search if you are not familiar with the theme, but, at the end got to the following:
- "protein structure prediction"
- "protein structure similarity"
- "protein structure modeling"
- protein "structure determination"
- automated "protein structure determination"
And decided to search in Google Scholar and PubMed.
Search throught does bases are easy and one could easily apply some filters, and that's was done. I'm limit my research filtering Full Papers from 2016 to 2020, getting the first 20 papers from best match results (the sites gives you the option to sort by date or best match).
After collect all papers, a new filter was made, preprocessing the papers by reading their abstract and seeing if there is some relationship with the theme, and the selected the most recently or the ones that have some interesting content. At the end I ended up with 22 papers to read.
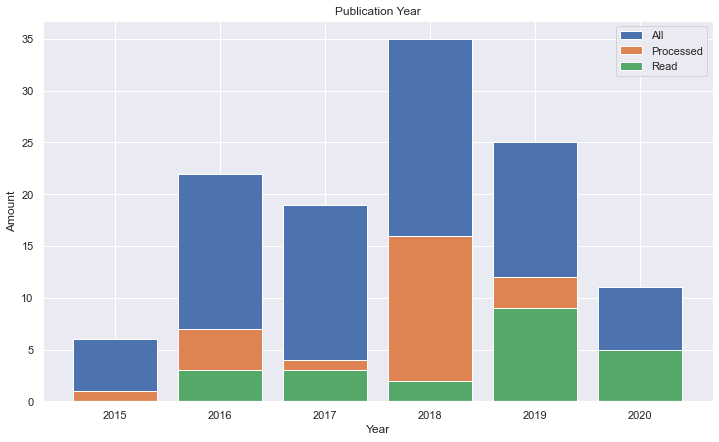
It's interesting to note that the number of papers in the research was getting high, but some strange down in 2019, so I thought why not to sum all papers from 2000 to 2020 given the terms.
Since PubMed gives a nice csv file to download, I joined the values for each of the terms over the years.
We could see a nice linear trend over the years.
Proteins structure
Protein structure are divided in four parts:
- Primary
- Secondary
- Tertiary
- Quaternary
I'm not going to describe each of the structure, but the image bellow may do an excellent job in illustrating what each structure is.
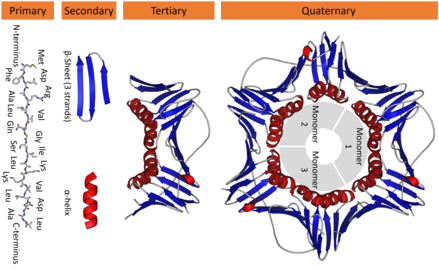
The important ones here (to understand this post) are the primary structure (which is the amino acid chain) and the tertiary structure (the 3D representation of the protein).
Prediction Models
After reading the papers (and search over the internet for terms and better explanation about topics related to proteins), there is a separation between two class of methods to predict protein structures:
"If the query protein has a homolog of known structure, the task is relatively easy and high-resolution models can often be built by copying and refining the framework of the solved structure. However, a template-based modeling procedure does not help answer the questions of how and why a protein adopts its specific structure. In particular, if structural homologs do not exist, or exist but cannot be identified, models have to be constructed from scratch. This procedure, called ab initio modeling, is essential for a complete solution to the protein structure prediction problem; it can also help us understand the physicochemical principle of how proteins fold in nature. Currently, the accuracy of ab initio modeling is low and the success is generally limited to small proteins (<120 residues)."1
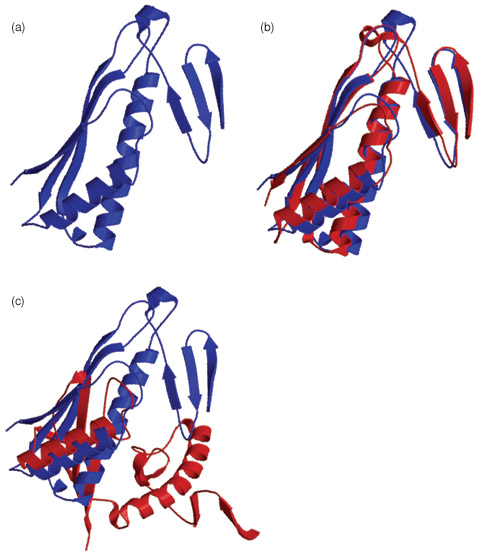
Above is a examplo from CASP6 of the original TM0919 protein (a), and a good (b) and not so good (c) prediction of this protein.
ab initio (template-free)
Methods in this class, seek to predict and build the tree-dimensional protein from scratch, taking into account physical restrictions.
From scratch means taking the amino acid sequence (the primary structure) and predict the tertiary structure.
One way of doing this prediction is to predict the contact map (actually the distance) between the amino acid s.
"Residue-residue contacts (or simply "contacts") in protein 3-D structures are pairs of spatially close residues. A 3-D structure of a protein is expressed as x, y, and z coordinates of the amino acids’ atoms in the form of a pdb file,1 and hence, contacts can be defined using a distance threshold. A pair of amino acids are in contact if the distance between their specific atoms (mostly carbon-alpha or carbon-beta) is less than a distance threshold (usually 8Å)..."2
This method, predicting the contact map is the one that Google's AlphaFold algorithm use, and is particularly well suited for deep learning methods given that the contact map is a 2D image.
Bellow is the example from Deep Minds excellent blog post3 about AlphaFold.
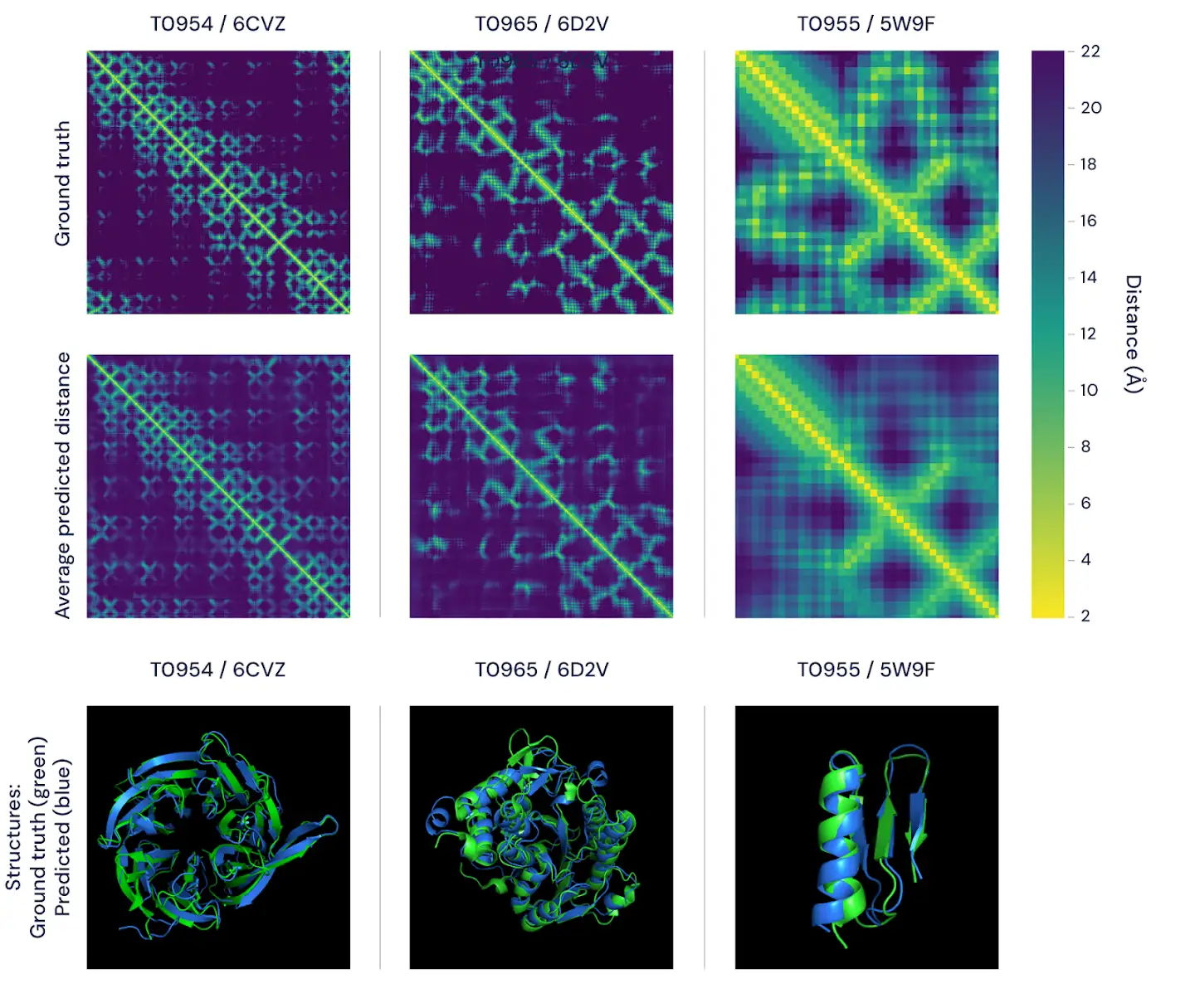
"Figure 2: Two ways of visualising the accuracy of alphafold’s predictions. the top figure features the distance matrices for three proteins. the brightness of each pixel represents the distance between the amino acids in the sequence comprising the protein–the brighter the pixel, the closer the pair. shown in the top row are the real, experimentally determined distances and, in the bottom row, the average of alphafold's predicted distance distributions. importantly, these match well on both global and local scales. the bottom panels represent the same comparison using 3d models, featuring alphafold’s predictions (blue) versus ground-truth data (green) for the same three proteins."3
Models and Tools:
template-based
The other approuch is know as template-based, and as the name suggest, the new proteins are predict based on already know structures. The models that use template-based approuch are the most precise to date, but it suffer from small number of proteins that already have been indentified by physical methods like X-ray crystallography, NMR spectroscopy and cryo-electron microscopy (also know as cryo-EM). Not that the ab initio doesn't use data from those sources, but template-based approuch need this to search the database for similar structures (so it's fully dependent).
Models that implement template-based can be derived in:
- Homology modeling
- Threading
Homology modeling in more dependent of protein databases than Threading, which uses a more statistical approuch, between proteins that exist in the database and the sequences of the one searching to solve.
Models and Tools:
Databases
Both predicting approuchs leaves us the need to have databases with protein strutures.
Over the years (the field is not new, but not too old either), a diverse number of databases were created. Bellow, there is a list of databases identified in the papers I've read (this is not a extensive list).
- DisProt: a database of intrinsically disordered proteins.
- HOMSTRAD: a database of protein structure alignments for homologous families
- MALIDUP: a database of pairwise, structure-base alignments for homologous domains originated from internal duplication.
- MobiDB: a database of protein disorder and mobility annotations
- ModBase: a database of comparative protein structure models
- MALISAM: a database of pairwise, structure-based alignments for structurally analogous motifs in proteins.
- UniProtKB: central hub for the collection of functional information on proteins, with accurate, consistent and rich annotation.
- wwPDB: Protein Data Bank
- RCSB PDB: Protein Data Bank
The PDB, is a large repository of proteins, and lot's of papers use their data to train models presented in in CASP (which we'll discuss later).
But, as mention before, the pace of protein discovery and availability in databases like PDB are getting higher, but not higher enough. Bellow bar chart presents the availiability of proteins in RCSB PDB over the years4.
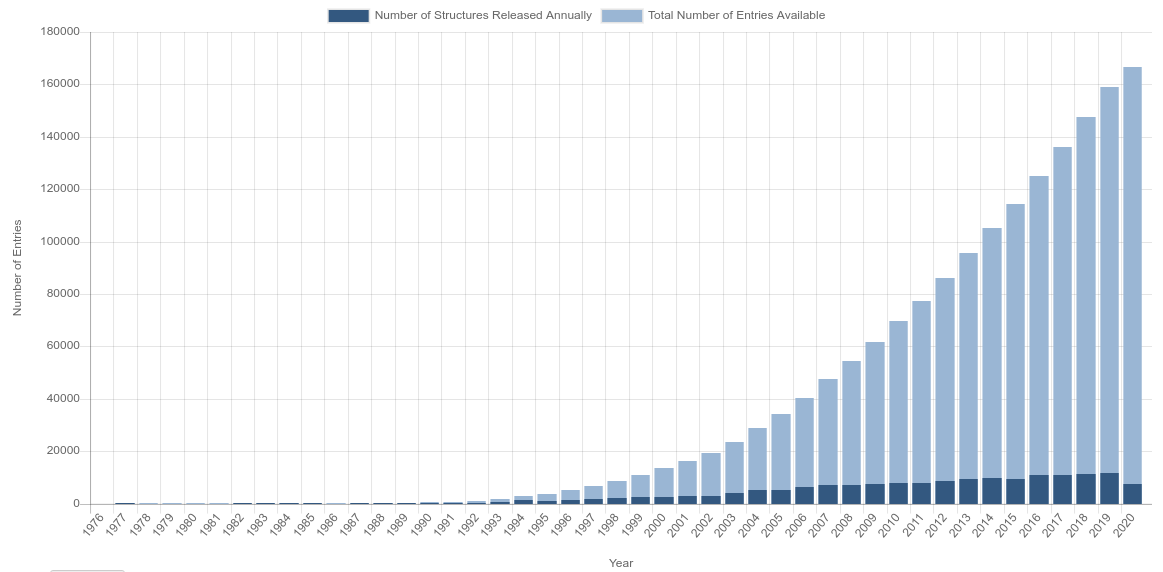
In the PDB the information about proteins are recorded in different file formats, but the one used is the .pdb, which contains information about the position of the atoms that make up the protein structure.
Another file format is FASTA, and the following in the amino acid sequence of 1BTT, is a "muscular" (signal transduction protein) protein found in House Mouse (Mus musculus), and the protein that is illustrated in the cover of this post.
>1BTN_1|Chain A|BETA-SPECTRIN|Mus musculus (10090)
MEGFLNRKHEWEAHNKKASSRSWHNVYCVINNQEMGFYKDAKSAASGIPYHSEVPVSLKEAICEVALDYKKKKHVFKLRLSDGNEYLFQAKDDEEMNTWIQAISSA
CASP (Critical Assessment of protein Structure Prediction)
All this journey, leaves us in CASP, which is a biennial event, whose "experiments aim at establishing the current state of the art in protein structure prediction, identifying what progress has been made, and highlighting where future effort may be most productively focused."5
The first CASP (CASP1) started in 1994 and now, in 2020, we should have CASP14. Google presented AlphaFold in CASP13 (two years ago), so the new edition promises better models and perhaps new findings. The graphics bellow demonstrat how good the CASP13 was in terms of improvement for the area.
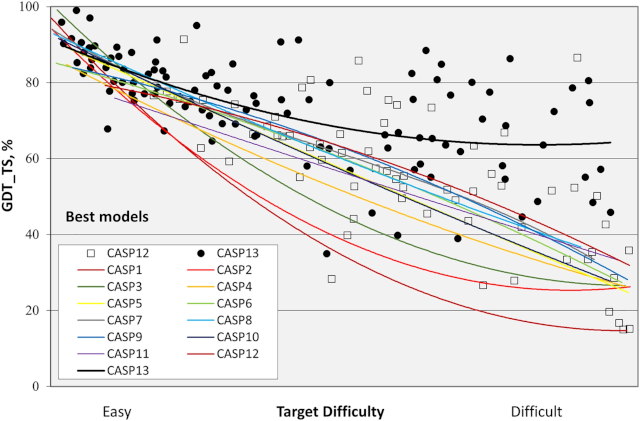
But, because of COVID-19, CASP14 has been delayed, so some results of the biennal event may appear only at the end of 2020, or maybe in 2021.
Something that wasn’t discussed in this post were other techniques and metrics to help predict protein structures (you could see in the image above one metric used to verify how good a model is, called GDT_TS).
So, if you like to know more about those other techniques and algorithms, i'm putting bellow some of them and you could search on Google to know more (some of them have their own Wikipedia page already). Some of them are used in CASP to evaluate the model, and other are used "inside" the models.
Structural Alignment:
- TMAlign
- FTAlign
- DeepAlign
- MADOKA
- GRAlign
- (3D) Yau-Hausdorff distance
Metrics for measuring the similarity, distance or quality:
- RMSD (root-mean-square deviation)
- TM-score
- GDT_TS
- CAD-score
- VoroMQA
Final Considerations
One thing I saw in the readings and perhaps it is a sign of movement is the use of machine learning (I believe that it was not initiated by Google, but perhaps fostered by AlphaFold), especially in ab initio models. In addition to these, hybrid models may also be implemented due to data scarcity.
However, this scarcity of data has a tendency to decrease due to the new cryo-EM technique, which promises to increase the amount of proteins identified, thus supplying databases like PDB with new data.
Finally, with the success of machine learning models, it may be that the area of bioinformatics will gain a new lease of life and be brought into focus as it had been a few years ago, allowing a new range of scientists to explore new possibilities.
References
1. Lee, Freddolino, and Zhang, Ab Initio Protein Structure Prediction.
2. Adhikari and Cheng, Protein Residue Contacts and Prediction Methods